1 思维导图

https://wiki.fysik.dtu.dk/gpaw/documentation/basic.html#manual-eigensolver
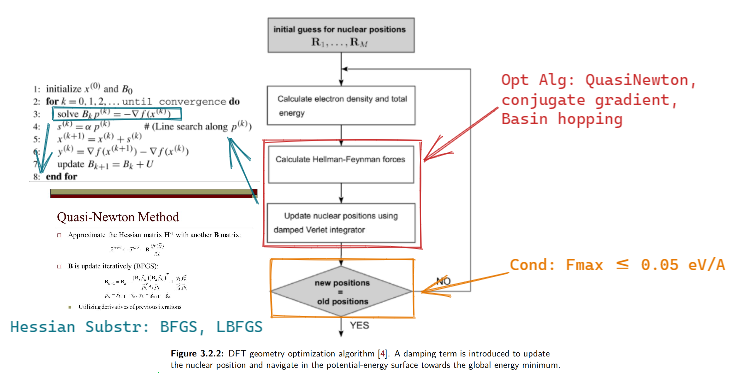
https://wiki.fysik.dtu.dk/ase/ase/optimize.html#module-ase.optimize

如何在Python中利用GPAW进行简单的DFT操作How to use GPAW to perform basic DFT tasks in Python_哔哩哔哩_bilibili
GPAW: DFT and beyond within the projector-augmented wave method_哔哩哔哩_bilibili
2 初始Image设置
2.1 初始结构准备
可以先进行分子动力学来进行预优化!
2.1.1 读取Slab
可以在VESTA中设定晶格常数(空间群)和原子位置,导出为合适的格式即可。用
slab = read('XXX.cif')`
2.1.2 吸附原子优化
from ase import Atoms
from ase.optimize import BFGS
from gpaw import GPAW
Mg2S2 = Atoms('Mg2S2',[[0,0,0],[3.7,0,0],[1.85,1.85,0],[1.85,-1.85,0]])
Mg2S2.center(vacuum=3.0) # x,y,z方向各加3 A的真空层
Mg2S2.calc = GPAW(mode='lcao', basis='dzp', txt='gpaw.txt')
opt = BFGS(atoms, trajectory='Mg2S2.traj')
opt.run(fmax=0.05)
2.1.3 将Mg2S2附加到slab上
add_adsorbate(slab,Mg2S2,height=4.0,position=(2.383, 5.455))
2.2 初始结构优化 12
显然,晶格常数cell和原子位置postion都会对能量造成影响,晶格常数实际上固定了位于格点的原子间距。可以全部采用实验值或者部分采用实验值进行优化。对于吸附体系显然没有实验值,所以需要一起优化。使用
ucf = UnitCellFilter(slab)
qn = QuasiNewton(ucf)
或者
ecf = ExpCellFilter(atoms)
qn = QuasiNewton(ecf)
但是,一起优化可能耗时长,可以用set_constraints的FixAtoms命令固定某些原子。准牛顿算法进行几何优化。用ase.constraints.StrainFilter固定某些方向。
前期的粗略计算可以先固定碳原子和晶格常数,后期再考虑影响大不大。
print(slab.constraint)
c = FixAtoms(mask=[atom.symbol == 'C' for atom in atoms])
slab.set_constraint(c)
2.3 超参数设置3
初始优化尽量快速,Ecut采用默认340 eV,泛函采用PBE,优化算法用QuasiNewton == BFGSLineSearch。
默认的k点为gamma点,即kpts=(1,1,1),多取几个点计算时间就会多几倍(可以并行),总能量是多个k点能量的平均,如果k点取得越密,该平均值越能反应布里渊区的电子能量分布。由于z方向不具备周期性,所以z方向的k点设置为0。不同k值反映了不同方向传播的电子,由于感受的周期不同,所以具有不同的能量。
后期可以进行修改超参数Ecut、Kpts、Ediff等或者扩胞。
由于我们关心的是相对能量而非决定能量,所以不用太关心超参数优化到最好的值。
3 最终Image设置
- 按照上述步骤建立final_image(如何选择初始的位置?)
4 优化算法
- 建立中间图像
configs = [initial.copy() for i in range(8)] + [final] # 8 of initial and 1 of final images
- 添加计算器
for config in configs:
config.calc = GPAW() # config the calculator of every image, only the internal images (not the endpoints) need have calculators attached.
- 线性插值并用CI-NEB算法
band = NEB(configs)
band.interpolate() # linear interpolation
relax = QuasiNewton(band, trajectory='XXX.traj')
- 绘制图像
import matplotlib.pyplot as plt
e0 = initial.get_potential_energy()
E = [config.get_potential_energy() - e0 for config in configs]
plt.plot(E)
5 并行计算、io、时间估计、可视化
结构优化时间 = 单次SCF的时间 × N,N取决于算法的高效性。因为下一次SCF取决于上一次SCF的结果,所以无法有效进行并行计算。
CI-NEB时间 = 结构优化时间 x Image数量。每个Image可以各自进行结构优化,所以可以并行。
!mpiexec -np 4 python3 xxx.py
write('xxx.traj', slab)
write('xxx.cif', slab)
!ase -gui xxx.traj/cif
将cif文件拖入http://www.openmx-square.org/viewer/
6 问题
- 具体的能量值代表什么?如何考虑费米狄拉克分布
width=0.1000 eV? - 实空间间隔如何取的?具体的能带数量如何通过Ecut求出?